The Technical Documentation must prove that the product fulfils the requirements laid down in the Medical Devices Directive
93/42/EEC. The
Technical File for medical devices should take account of the following aspects:
- Table of Contents with revision status and proof of completeness of the file
- Name and address of the manufacturer and/or if necessary: name of the European representative also and all places of manufacturing, excerpt from the commercial register or a registration of a business
- Description of product family including all variations planned and the Intended Use
- Declaration of Conformity
- Proof of fulfillment of the Essential Requirements according to Annex I
- Risk analysis in accordance with EN ISO 14971:2007
- List of the applied standards
- Information for use (e.g. Instructions for Use, Technical description)
- Product identification (e.g. labels, warnings, used symbols)
- Description of Packaging and Labeling
- If applicable, design and production drawings as well as plans of mechanical components and devices ect. (mechanical drawings) and Electrical Circuits (Block Diagram)
- If applicable, results of design calculations and performed testing
- If applicable, used method and validation report of sterilization Biocompatibility
- If applicable, test reports of electromagnetic compatibility (EN 60601-1-2:2007)
- Clinical evaluation according to Annex X, MDD
Simply speak to us – we look forward to hearing from you!
As a manufacturer, you may only market medical devices in the European market if all the requirements of the relevant Directive have been complied with. As one of the few Notified Bodies for all the Directives listed on the left, we help you carry out your conformity assessment procedure for CE marking as well as assist you to acquire worldwide approval of your medical devices.
- Definition of the medical device/intended use
As a manufacturer, you determine the intended use of your device. This results in a decision as to which directive is applicable to your medical device (MDD, AIMD or IVDD).
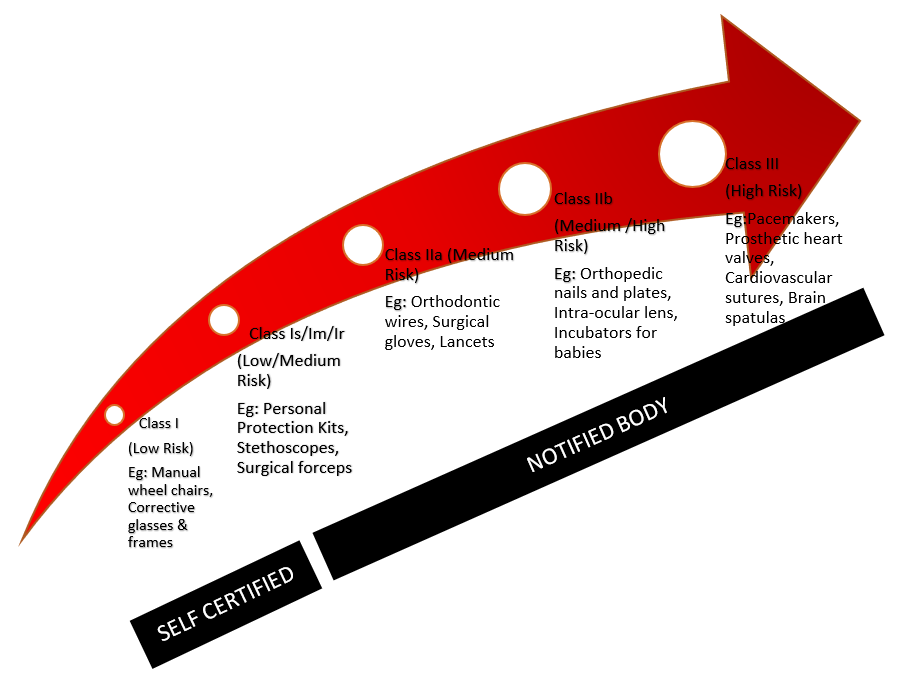
- Classification of the medical device
In the MDD, the medical device is classified in accordance with the regulations in Annex IX. The AIMD does not provide for any classification. The IVDD distinguishes between products for self-testing, for performance evaluation, Lists A and B and products which do not fall into any of the categories named.
- Determining the suitable procedure for conformity assessment
Depending on the classification of the product, a Notified Body is required which, for example, you will entrust with carrying out an EC type-examination, with assessing a design dossier and/or with auditing your QM system. AIMD products always require the involvement of a Notified Body.
The manufacturer must prepare a technical documentation for every type of medical device, which essentially consists of the following documentation on the product:
Description of the device, design documents, the standards to be applied and a description of the solutions chosen to meet the essential requirements, risk analysis, test reports, clinical data, labelling and instructions for use, and additionally for sterile devices descriptions of the sterilization procedures used and the validation certificates.
Medical devices must meet the applicable essential requirements listed in Annex I of the respective directive. With the help of the conformity assessment procedure, proof must be provided that the safety requirements have been met and that the technical services have been rendered. The medical effectiveness must be proven through a clinical assessment. The manufacturer is free to choose how he will provide the proof that the essential requirements have been met.
As a test body for medical devices, we can carry out various tests for you, such as safety, functional and biocompatibility tests. We perform them either in our own laboratories or in contract laboratories. You can use the resulting test reports as an acknowledged proof for your
MDR technical file .
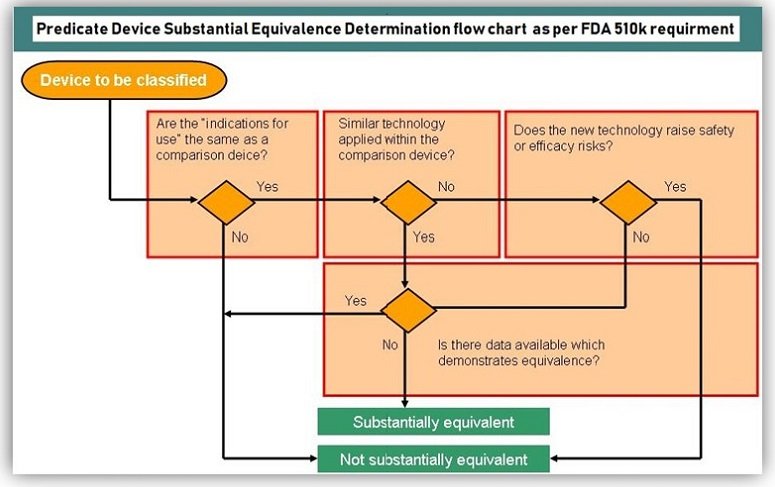
- EC type-examination or examination of the design of the product
For all AIMD, MDD Class III and IVDD List A products, the manufacturer must decide between the EC type-examination procedure and the examination of the product design documentation (design dossier). In this process, we -as your Notified Body- either test the product including the product documentation or the design dossier. After successful examination, our certification body issues an approval in accordance with the directive.
- Implementing the conformity assessment procedure
Usually, the manufacturers of medical devices choose the procedure of auditing their quality management system. The auditors assess the documentation of the QM system prior to the audit and verify the application of the written procedures during the audit at the company. The duration of the audit and the number of auditors depend on the size of the company and the variety and kinds of products and are specified in an individual quotation. After the audit, you will receive a written report with the audit results.
As an alternative to the audit, the EC verification procedure (sampling or individual testing) in accordance with Annex IV (MDD) or Annex VI (IVDD) can be applied. If the conformity assessment procedure has been completed successfully, our certification body will issue an approval for the CE marking of your devices.
Simply speak to us – we look forward to hearing from you!
Conformity Assessment Procedure
Handling processes can be speeded up. A first step is our following sequence plan (for medical devices in accordance with the AIMD, MDD, IVDD directives):
First, we present ourselves and our range of services and get to know you, your company and your products. We then determine further procedures together. Early establishing of communication, e.g. as early as in the product development phase, allows for better coordination of the time schedule for the market launch of your product.
We will then prepare a quotation for you covering the services you request on the way to acquiring CE marking (QM audit according to
EN ISO 13485 and
EN ISO 9001, product testing, examination of the technical documentation, etc.).
After receipt of the order, we will discuss any open questions and coordinate the project handling together with you.
Prior to the audit, selected documents out of the technical documentation will be checked for plausibility and for conformity with the requirements of the directive.
- For all AIMD, MDD Class III and IVDD List A products
Examination of the design dossier or EC type-examination. You submit to us the documentation on the product design (design dossier), and we will check it for completeness and plausibility. Or we test your product and the associated documentation. We will then issue a report on the test results. If the assessment is positive, you will receive the following certificates:
We will base the scope of the pre audit on your wishes. You will receive an audit report in which potential for improvements will be pointed out.
- Review of the QM documentation
The QM documents, such as the Quality Manual as well as relevant procedures, will be assessed by us prior to the audit, and you will receive a report on our findings.
Prior to the audit, you will receive an audit plan agreed on with you. During the audit, the operations in your company will be examined and thus the implementation and effectiveness of the requirements of the applicable directive and the relevant standards. In the process, we will particularly assess the conformity of the products with the essential requirements. The team of auditors always includes at least one expert on your product range. All relevant production sites are included in the scope of the audit. You will receive a detailed report on the performed audit that includes also advice for improvement.
If the audit result is positive, you will receive an approval for the selected conformity assessment procedure.
In addition, certificates according to QM standards EN ISO
13485 and where required EN
ISO 9001 are issued. Afterwards, we will carry out surveillance audits every twelve months and a recertification audit after five years.
- Declaration of Conformity
You are now permitted to issue the declaration of conformity for your medical devices, to provide them with the CE marking including the identification number of the “Notified Body” and to place them on the European market.
Contact us – we are looking forward to hearing from you!