The classification of medical devices in the European Union is outlined in Annex IX of the Council Directive 93/42/EEC. There are basically four classes, ranging from low risk to high risk.
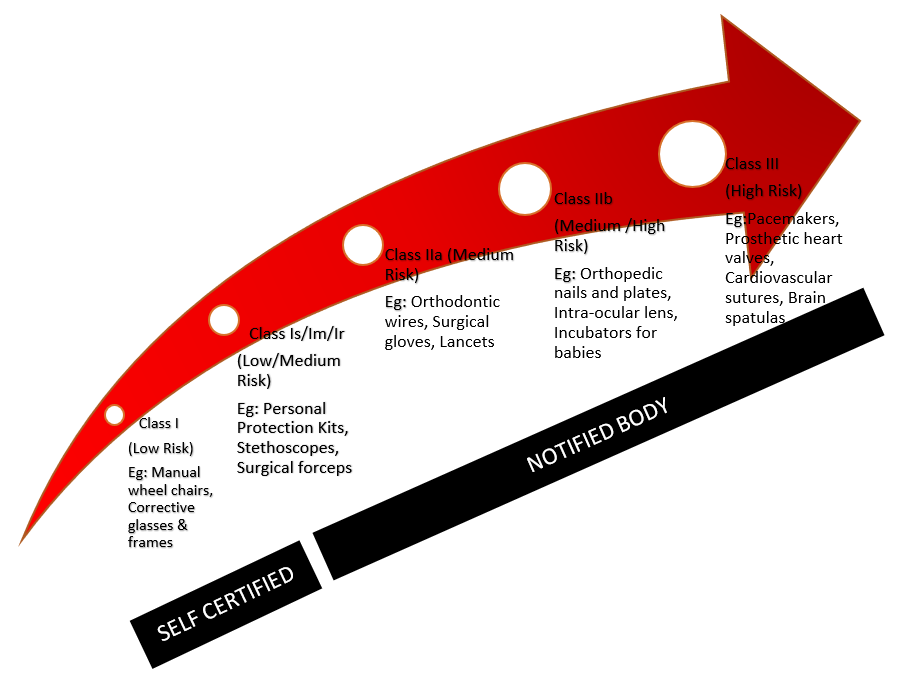
A medical device (MD) may be classified as Class 1**(including Is & Im), IIa**, IIb** and III**, with Class III covering the highest risk products. The higher the classification the greater the level of assessment required. All Class Is, Im, IIa, IIb and III medical devices require the intervention of third party: the so-called Notified Body Medical Device Classification .
The classification rules are set out in Annex IX of the directive. This annex includes definitions of the terminology used in the classification rules. Classification of a medical device will depend up on a series of factors, including:
- how long the device is intended to be in continuous use
- whether or not the device is invasive or surgically invasive,
- whether the device is implantable or active
- whether or not the device contains a substance, which in its own right is considered to be a medicinal substance and has action ancillary to that of the device.
The authorization of medical devices is guaranteed by a Declaration of Conformity. (FAQ)This declaration is issued by the manufacturer itself, but for products in Class Is, Im, IIa, IIb or III, it must be verified by a Certificate of Conformity issued by a Notified Body. (FAQ) Medical devices that pertain to class I (on condition they do not need to be sterilized or are not used to measure a function) can be put on the market purely by self-certification.
Certified medical devices should have the CE mark on the packaging, insert leaflets, etc. These packaging should also show harmonized pictograms and EN standardized logos to indicate essential features such as instructions for use, expiry date, manufacturer, sterile, don’t reuse, etc.